Da Real Adogg Sen. Kennedy GRILLS Leftist For Saying GANG MEMBERS Shouldn’t Be ARRESTED For Having Illegal Guns April 25, 2024 0
Black Conservative Perspective trump Trump Deranged Ex-NFL Player HOSTS CRINGE VENTING SESSION Over Black People Supporting Trump! April 25, 2024 0
Middle MAGA WATCH LIVE: LAPD dispersing protesters at USC – BREAKING NEWS (College Protests) April 24, 2024 0
Black Conservative Perspective WOKE College Graduate BREAKS DOWN On Camera After Real Life Reality Check That Her Degree Is USELESS April 24, 2024 0
Man Guide #1 Reason Why 99.9% Western Women Are UNDATEABLE These Days…Can’t Handle Men Have Own Standards… April 24, 2024 0
Middle MAGA SHOCKING DEBATE: Tucker Carlson Calls Atomic Bombing ‘EVIL’; Ben Shapiro ARGUES It Saved Lives *clip April 24, 2024 0
biden Black Conservative Perspective Charles Payne SNAPS On Democrat Apologist Defending Joe Biden’s INSANE Mental Breakdowns! April 24, 2024 0
O’Keefe Media Group Styxhexenhammer666 OMG Exclusive with James O’Keefe featuring Erik Prince April 24, 2024 0
Timcast Amazon & Wal Mart ABANDON Self Check Out, Skyrocketing Crime Causes Companies To bring Back HUMANS April 24, 2024 0
Da Real Adogg trump 🚨Judge Cannon Sides With TRUMP & FINALLY REMOVES Jack Smith After She OPEN His UNREDACTED DOCUMENTS April 24, 2024 0
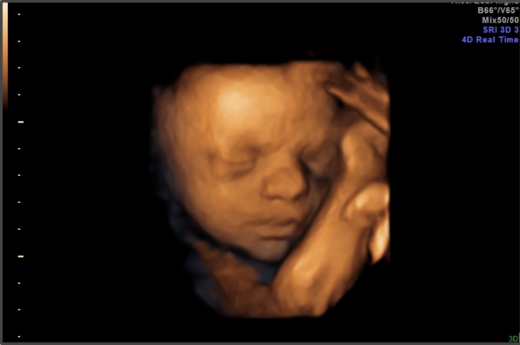
Home Posts LifeNews Opinion Ultrasounds Remind us That Unborn Babies are Human Beings April 24, 2024 0